
เนื้อหา iLive ทั้งหมดได้รับการตรวจสอบทางการแพทย์หรือตรวจสอบข้อเท็จจริงเพื่อให้แน่ใจว่ามีความถูกต้องตามจริงมากที่สุดเท่าที่จะเป็นไปได้
เรามีแนวทางการจัดหาที่เข้มงวดและมีการเชื่อมโยงไปยังเว็บไซต์สื่อที่มีชื่อเสียงสถาบันการวิจัยทางวิชาการและเมื่อใดก็ตามที่เป็นไปได้ โปรดทราบว่าตัวเลขในวงเล็บ ([1], [2], ฯลฯ ) เป็นลิงก์ที่คลิกได้เพื่อการศึกษาเหล่านี้
หากคุณรู้สึกว่าเนื้อหาใด ๆ ของเราไม่ถูกต้องล้าสมัยหรือมีข้อสงสัยอื่น ๆ โปรดเลือกแล้วกด Ctrl + Enter
โรคชาร์คอต-มารี-ทูธ
ผู้เชี่ยวชาญทางการแพทย์ของบทความ

ตรวจสอบล่าสุด: 12.07.2025
โรคกล้ามเนื้อฝ่อด้านข้างหรือกลุ่มอาการ Charcot-Marie-Tooth เป็นกลุ่มโรคเรื้อรังทางพันธุกรรมที่ทำให้เส้นประสาทส่วนปลายได้รับความเสียหาย
ตาม ICD-10 ในส่วนของโรคระบบประสาท รหัสของโรคนี้คือ G60.0 (โรคระบบประสาทสั่งการและประสาทรับความรู้สึกที่ถ่ายทอดทางพันธุกรรม) ซึ่งรวมอยู่ในรายชื่อโรคกำพร้าด้วย
ระบาดวิทยา
ตามสถิติทางคลินิก พบว่าอัตราการเกิดโรค Charcot-Marie-Tooth ทุกประเภทต่อประชากร 100,000 คนอยู่ที่ 19 ราย (ตามแหล่งข้อมูลอื่นๆ พบว่ามี 1 รายต่อประชากร 2,500-10,000 คน)
CMT ชนิดที่ 1 คิดเป็นประมาณสองในสามของผู้ป่วยทั้งหมด (หนึ่งรายต่อประชากร 5,000 ถึง 7,000 คน) และเกือบ 70% ของผู้ป่วยเหล่านี้มีความเกี่ยวข้องกับการซ้ำซ้อนของยีน PMP22 ผู้คนมากกว่า 1.2 ล้านคนทั่วโลกต้องทนทุกข์ทรมานจากโรคประเภทนี้
อุบัติการณ์ของ CMT ชนิดที่ 4 คาดว่าอยู่ที่ 1-5 รายต่อเด็ก 10,000 คน [ 1 ]
สาเหตุ โรคชาร์คอต-มารี-ทูธ
ตามการจำแนกประเภทของกลุ่มอาการโพลีนิวโรพาธีกล้ามเนื้อฝ่อของ peroneal (fibular) กล้ามเนื้ออ่อนแรงของเส้นประสาท Charcot-Marie-Tooth หรือโรค Charcot-Marie-Tooth (ย่อว่า CMT) หมายถึงโพลีนิวโรพาธีที่รับความรู้สึกทางการเคลื่อนไหวซึ่งกำหนดโดยพันธุกรรม [ 2 ]
นั่นคือ สาเหตุของการเกิดขึ้นคือการกลายพันธุ์ทางพันธุกรรม และขึ้นอยู่กับลักษณะของการเบี่ยงเบนทางพันธุกรรม ประเภทหรือชนิดของกลุ่มอาการนี้หลักๆ จะแตกต่างกัน คือ ไมอีลินเสื่อมและแอกซอน กลุ่มแรกได้แก่ Charcot-Marie-Tooth disease type 1 (CMT1) ซึ่งเกิดจากการซ้ำซ้อนของยีน PMP22 บนโครโมโซม 17 ซึ่งเข้ารหัสโปรตีนไมอีลินรอบนอก 22 ข้ามเยื่อหุ้มเซลล์ เป็นผลให้เกิดการเสื่อมของไมอีลินแบบแบ่งส่วนของปลอกแอกซอน (กระบวนการของเซลล์ประสาท) และความเร็วในการนำสัญญาณประสาทลดลง นอกจากนี้ การกลายพันธุ์อาจเกิดขึ้นในยีนอื่นๆ อีกด้วย
รูปแบบแอกซอนคือ Charcot-Marie-Tooth disease type 2 (CMT2) ซึ่งส่งผลต่อแอกซอนเองและเกี่ยวข้องกับการเปลี่ยนแปลงทางพยาธิวิทยาในยีน MFN2 ที่ตำแหน่ง 1p36.22 ซึ่งเข้ารหัสโปรตีนเยื่อหุ้มเซลล์ไมโตฟูซิน-2 ซึ่งจำเป็นสำหรับการหลอมรวมไมโตคอนเดรียและการสร้างเครือข่ายไมโตคอนเดรียที่ทำงานได้ภายในเซลล์ประสาทส่วนปลาย มี CMT2 มากกว่าสิบชนิดย่อย (โดยมีการกลายพันธุ์ในยีนเฉพาะ)
ควรสังเกตว่าปัจจุบันมีการระบุยีนมากกว่าร้อยยีนซึ่งความเสียหายที่ถ่ายทอดทางกรรมพันธุ์ทำให้เกิดโรค Charcot-Marie-Tooth หลายประเภท ตัวอย่างเช่น การกลายพันธุ์ในยีน RAB7 ทำให้เกิด CMT ประเภท 2B การเปลี่ยนแปลงของยีน SH3TC2 (ซึ่งเข้ารหัสโปรตีนเยื่อหุ้มเซลล์ชนิดหนึ่งของเซลล์ชวานน์) ทำให้เกิด CMT ประเภท 4C ซึ่งแสดงอาการในวัยเด็กและมีลักษณะเฉพาะคือไมอีลินในเซลล์ประสาทสั่งการและเซลล์ประสาทรับความรู้สึกถูกทำลาย (โรคประเภท 4 นี้มีประมาณสิบกว่าแบบ)
CMT ชนิด 3 ที่หายาก (เรียกว่ากลุ่มอาการ Dejerine-Sottas) เริ่มพัฒนาในวัยเด็ก และเกิดจากการกลายพันธุ์ใน PMP22, MPZ, EGR2 และยีนอื่นๆ
เมื่อ CMT ชนิดที่ 5 เกิดขึ้นในวัย 5-12 ปี ไม่เพียงแต่จะสังเกตเห็นอาการเส้นประสาทสั่งการการเคลื่อนไหว (ในรูปแบบของอัมพาตครึ่งล่างของแขนหรือขาแบบเกร็ง) แต่ยังพบความเสียหายของเส้นประสาทตาและเส้นประสาทการได้ยินอีกด้วย
อาการกล้ามเนื้ออ่อนแรงและการฝ่อของเส้นประสาทตา (ร่วมกับการสูญเสียการมองเห็น) เช่นเดียวกับปัญหาการทรงตัว ถือเป็นลักษณะเฉพาะของผู้ป่วย CMT ประเภท 6 ส่วนโรค Charcot-Marie-Tooth ประเภท 7 ไม่เพียงแต่มีโรคเส้นประสาทรับความรู้สึกด้านการเคลื่อนไหวเท่านั้น แต่ยังมีอาการโรคจอประสาทตาในรูปแบบของเรตินิติสพิกเมนโตซาด้วย
โรค CMT ที่เชื่อมโยงกับโครโมโซม X หรือโรค Charcot-Marie-Tooth ที่พบได้บ่อยในผู้ชาย โดยมีอาการแขนขาอ่อนแรงทั้ง 4 ข้าง (แขนและขาอ่อนแรง) เป็นโรคที่ทำให้ไมอีลินเสื่อมลง และเชื่อว่าเกิดจากการกลายพันธุ์ของยีน GJB1 บนแขนยาวของโครโมโซม X ซึ่งเข้ารหัสคอนเน็กซิน 32 ซึ่งเป็นโปรตีนข้ามเยื่อหุ้มเซลล์ของเซลล์ชวานน์และโอลิโกเดนโดรไซต์ที่ควบคุมการส่งสัญญาณประสาท [ 3 ]
ปัจจัยเสี่ยง
ปัจจัยเสี่ยงหลักของ CMT คือประวัติครอบครัวที่เป็นโรคนี้ นั่นคือ ญาติสนิท
นักพันธุศาสตร์ระบุว่า หากทั้งพ่อและแม่เป็นพาหะของยีนถ่ายทอดทางพันธุกรรมแบบด้อยสำหรับโรค Charcot-Marie-Tooth ความเสี่ยงที่ลูกจะเป็นโรคนี้จะอยู่ที่ 25% และความเสี่ยงที่ลูกจะเป็นพาหะของยีนนี้ (แต่จะไม่มีอาการใดๆ) อยู่ที่ประมาณ 50%
ในกรณีของการถ่ายทอดทางพันธุกรรมแบบ X-linked (เมื่อยีนที่กลายพันธุ์อยู่บนโครโมโซม X ของผู้หญิง) มีความเสี่ยง 50% ที่แม่จะถ่ายทอดยีนดังกล่าวไปยังลูกชายซึ่งจะพัฒนาเป็น CMT โรคนี้อาจไม่เกิดขึ้นเมื่อมีทารกเพศหญิง แต่ลูกชาย (หลาน) ของลูกสาวอาจสืบทอดยีนที่ผิดปกติ และโรคก็จะเกิดขึ้น
กลไกการเกิดโรค
โรค Charcot-Marie-Tooth ทุกประเภท เกิดจากความผิดปกติทางพันธุกรรมของเส้นประสาทส่วนปลาย ได้แก่ ระบบประสาทสั่งการ (การเคลื่อนไหว) และระบบประสาทรับความรู้สึก (ความรู้สึกไว)
หาก CMT ชนิด demyelination การทำลายหรือการบกพร่องของปลอกไมอีลินที่ปกป้องแอกซอนของเส้นประสาทส่วนปลายจะทำให้การส่งผ่านกระแสประสาทไปยังระบบประสาทส่วนปลายระหว่างสมอง กล้ามเนื้อ และอวัยวะรับความรู้สึกช้าลง
ในกรณีของโรคชนิดแอกซอน แอกซอนเองจะได้รับผลกระทบ ทำให้ความแรงของสัญญาณประสาทลดลง ส่งผลให้สัญญาณประสาทไม่แรงพอที่จะกระตุ้นกล้ามเนื้อและอวัยวะรับความรู้สึกได้เต็มที่
อ่านเพิ่มเติม:
โรค Charcot-Marie-Tooth ถ่ายทอดได้อย่างไร ยีนที่ผิดปกติสามารถถ่ายทอดได้แบบถ่ายทอดทางพันธุกรรมแบบถ่ายทอดทางพันธุกรรมแบบเด่นหรือแบบถ่ายทอดทางพันธุกรรมแบบด้อย
ประเภทที่พบมากที่สุด คือ การถ่ายทอดทางพันธุกรรมแบบออโตโซมัลโดมิแนนต์ ซึ่งเกิดขึ้นเมื่อมียีนที่กลายพันธุ์เพียงชุดเดียว (ถ่ายทอดโดยพ่อแม่ฝ่ายใดฝ่ายหนึ่ง) และความน่าจะเป็นในการถ่ายทอด CMT ให้กับเด็กที่เกิดแต่ละคนนั้นประมาณอยู่ที่ 50% [ 4 ]
ในกรณีถ่ายทอดทางพันธุกรรมแบบถ่ายทอดทางพันธุกรรมลักษณะด้อย จำเป็นต้องมียีนที่ผิดปกติ 2 ชุด (ชุดละ 1 ชุดจากพ่อแม่ซึ่งไม่แสดงอาการใดๆ ของโรค) จึงจะเกิดการพัฒนาของโรคได้
ใน 40-50% ของกรณี พบว่ามีการทำลายไมอีลินทางพันธุกรรมแบบถ่ายทอดทางออโตโซมัลโดมิแนนต์ เช่น CMT ชนิดที่ 1 ใน 12-26% ของกรณี พบว่ามีการทำลายไมอีลินของแอกซอน เช่น ชนิดที่ 2 และใน 10-15% ของกรณี พบว่ามีการถ่ายทอดทางพันธุกรรมแบบ X-linked [ 5 ]
อาการ โรคชาร์คอต-มารี-ทูธ
โดยทั่วไปอาการเริ่มแรกของโรคนี้จะเริ่มปรากฏในวัยเด็กและวัยรุ่นและค่อยๆ พัฒนาขึ้นตลอดชีวิต แม้ว่ากลุ่มอาการอาจแสดงออกมาในภายหลังก็ตาม อาการต่างๆ ที่เกิดขึ้นนั้นไม่แน่นอน และไม่สามารถคาดเดาอัตราการดำเนินของโรค รวมถึงความรุนแรงของโรคได้
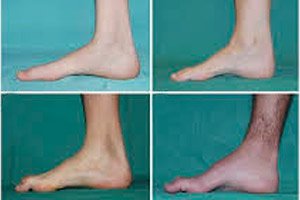
อาการทั่วไปในระยะเริ่มแรก ได้แก่ ความเหนื่อยล้าทั่วไปที่เพิ่มขึ้น กล้ามเนื้อของเท้า ข้อเท้า และหน้าแข้งอ่อนแรงลง ปฏิกิริยาตอบสนองลดลง ทำให้การเคลื่อนไหวของเท้าทำได้ยากและนำไปสู่อาการเดินผิดปกติ (dysbasia) โดยขาจะยกสูงขึ้น มักสะดุดและล้มบ่อยครั้ง อาการของโรค Charcot-Marie-Tooth ในเด็กเล็กอาจรวมถึงความเก้กังที่เด่นชัดและความยากลำบากในการเดินที่ผิดปกติตามวัยซึ่งเกี่ยวข้องกับ อาการ เท้าตกทั้งสองข้างนอกจากนี้ ความผิดปกติของเท้ายังมีลักษณะเฉพาะ ได้แก่ อุ้งเท้าสูง (เท้ากลวง) หรือเท้าแบนอย่างรุนแรง นิ้วเท้าโค้งงอ (รูปค้อน)
ในกรณีที่เดินโดยใช้ปลายเท้าโดยมีภาวะกล้ามเนื้ออ่อนแรง แพทย์ระบบประสาทอาจสงสัยว่าเด็กเป็น CMT ประเภทที่ 4 ซึ่งเด็กอาจไม่สามารถเดินได้เมื่อเข้าสู่วัยรุ่น
เมื่อโรคดำเนินไป กล้ามเนื้อจะฝ่อและอ่อนแรงลงและลามไปยังแขนขาส่วนบน ทำให้เคลื่อนไหวกล้ามเนื้อมัดเล็กได้ยากและทำกิจกรรมมือปกติได้น้อยลง การรับรู้ทางสัมผัสที่ลดลงและความสามารถในการรู้สึกถึงความร้อนและความเย็น รวมถึงอาการชาที่เท้าและมือ บ่งชี้ถึงความเสียหายของแอกซอนของเส้นประสาทรับความรู้สึก
ในโรค Charcot-Marie-Tooth ชนิดที่ 3 และ 6 ซึ่งแสดงอาการในวัยเด็ก จะมีอาการอะแท็กเซีย (การประสานงานการเคลื่อนไหวและการทรงตัวบกพร่อง) กล้ามเนื้อกระตุกและสั่น เส้นประสาทใบหน้าเสียหาย เส้นประสาทตาฝ่อร่วมกับอาการตาสั่น และสูญเสียการได้ยิน
ในระยะต่อมาอาจมีอาการสั่นอย่างควบคุมไม่ได้ (tremor) และเป็นตะคริวกล้ามเนื้อบ่อยครั้ง ปัญหาในการเคลื่อนไหวอาจนำไปสู่การเกิดอาการปวดกล้ามเนื้อ ข้อต่อ และเส้นประสาท
ภาวะแทรกซ้อนและผลกระทบ
โรค Charcot-Marie-Tooth อาจมีภาวะแทรกซ้อนและผลที่ตามมา เช่น:
- เกิดอาการเคล็ดขัดยอกหรือกระดูกหักบ่อยมากขึ้น
- การหดตัวที่เกี่ยวข้องกับการสั้นลงของกล้ามเนื้อและเอ็นรอบข้อ
- กระดูกสันหลังคด (กระดูกสันหลังคด)
- ปัญหาการหายใจ – เมื่อเส้นประสาทที่เลี้ยงกล้ามเนื้อกะบังลมได้รับความเสียหาย:
- การสูญเสียความสามารถในการเคลื่อนไหวได้อย่างอิสระ
การวินิจฉัย โรคชาร์คอต-มารี-ทูธ
การวินิจฉัยประกอบด้วยการตรวจร่างกาย การตรวจประวัติครอบครัว (รวมถึงประวัติครอบครัว) การตรวจระบบประสาทและการตรวจระบบร่างกาย
การทดสอบจะดำเนินการเพื่อตรวจสอบช่วงการเคลื่อนไหว ความไว และการตอบสนองของเอ็น การนำกระแสประสาทสามารถประเมินได้โดยใช้การวินิจฉัยด้วยเครื่องมือ เช่น การตรวจคลื่นไฟฟ้ากล้ามเนื้อหรือ การตรวจ คลื่นไฟฟ้าประสาทกล้ามเนื้อ อาจต้องใช้การตรวจด้วยคลื่นอัลตราซาวนด์หรือ MRI ด้วย [ 6 ]
การตรวจทางพันธุกรรมหรือ DNA เพื่อตรวจหาการกลายพันธุ์ทางพันธุกรรมที่พบบ่อยที่สุดซึ่งทำให้เกิด CMT ในตัวอย่างเลือดนั้นมีข้อจำกัด เนื่องจากปัจจุบันยังไม่มีการทดสอบ DNA สำหรับโรคทุกประเภท สำหรับข้อมูลเพิ่มเติม โปรดดูที่การตรวจทางพันธุกรรม
ในบางกรณีจะมีการตรวจชิ้นเนื้อเส้นประสาทส่วนปลาย (โดยทั่วไปคือเส้นประสาทน่อง)
การวินิจฉัยที่แตกต่างกัน
การวินิจฉัยแยกโรครวมถึงโรคเส้นประสาทส่วนปลายอื่นๆ โรคกล้ามเนื้ออ่อนแรงแบบดูเชนน์ กลุ่มอาการไมอีโลพาธิกและไมแอสทีนิก โรคเส้นประสาทจากเบาหวาน โรคไมอีโลพาธิกในโรคปลอกประสาทเสื่อมแข็งและกล้ามเนื้ออ่อนแรง กลุ่มอาการกิลแลง-บาร์เร การบาดเจ็บที่เส้นประสาทบริเวณหน้าแข้งและการฝ่อ (รวมถึงเมื่อถูกกดทับระหว่างหมอนรองกระดูกสันหลังส่วนเอว) ความเสียหายที่สมองน้อยหรือทาลามัส รวมถึงผลข้างเคียงของเคมีบำบัด (ในระหว่างการรักษาด้วยยารักษาแบบไซโทสแตติก เช่น วินคริสตินหรือแพคลิแท็กเซล) [ 7 ]
ใครจะติดต่อได้บ้าง?
การรักษา โรคชาร์คอต-มารี-ทูธ
ปัจจุบันการรักษาโรคทางพันธุกรรมนี้ได้แก่ การออกกำลังกาย (เพื่อเพิ่มความแข็งแรงและยืดกล้ามเนื้อ) การบำบัดด้วยการทำงาน (ซึ่งช่วยผู้ป่วยที่มีกล้ามเนื้ออ่อนแรงที่มือ) และการใช้เครื่องช่วยพยุงร่างกายเพื่อช่วยในการเดิน หากจำเป็น แพทย์จะสั่งจ่ายยาแก้ปวดหรือยากันชัก [ 8 ]
ในกรณีของภาวะเท้าแบนอย่างรุนแรง สามารถทำการผ่าตัดกระดูกได้ และในกรณีที่มีส้นเท้าผิดรูป แนะนำให้ใช้วิธีแก้ไขด้วยการผ่าตัด – การผ่าตัดข้อ [ 9 ]
ขณะนี้มีการวิจัยเกี่ยวกับองค์ประกอบทางพันธุกรรมของโรคและวิธีการรักษาอยู่ การใช้เซลล์ต้นกำเนิด ฮอร์โมนบางชนิด เลซิติน หรือกรดแอสคอร์บิกยังไม่ได้รับผลเชิงบวก
อย่างไรก็ตาม จากการวิจัยล่าสุด อาจมีการค้นพบสิ่งใหม่ ๆ เกิดขึ้นเกี่ยวกับการรักษาโรค Charcot-Marie-Tooth ในอนาคตอันใกล้นี้ บริษัท Pharnext ของฝรั่งเศสจึงได้พัฒนายานี้มาตั้งแต่ปี 2014 และตั้งแต่กลางปี 2019 การทดลองทางคลินิกสำหรับยา PXT3003 เพื่อรักษา CMT ชนิดที่ 1 ในผู้ใหญ่ ซึ่งยับยั้งการแสดงออกของยีน PMP22 ที่เพิ่มขึ้น ปรับปรุงการสร้างไมอีลินของเส้นประสาทส่วนปลาย และบรรเทาอาการทางระบบประสาทและกล้ามเนื้อ
ผู้เชี่ยวชาญจากบริษัทการแพทย์ Sarepta Therapeutics (สหรัฐอเมริกา) กำลังดำเนินการสร้างยีนบำบัดสำหรับโรค Charcot-Marie-Tooth ชนิดที่ 1 การบำบัดนี้จะใช้ไวรัสที่เกี่ยวข้องกับอะดีโน (AAV) ที่ไม่เป็นอันตรายในสกุล Dependovirus ที่มีจีโนม DNA สายเดี่ยวเชิงเส้น ซึ่งจะถ่ายโอนยีน NTF3 เข้าสู่ร่างกายเพื่อเข้ารหัสโปรตีนนิวโรโทรฟิน-3 (NT-3) ซึ่งจำเป็นต่อการทำงานของเซลล์ประสาทชวานน์
Helixmith จะเริ่มการทดลองทางคลินิกของยีนบำบัด Engensis (VM202) ที่พัฒนาโดยเกาหลีใต้ภายในสิ้นปี 2020 เพื่อรักษาอาการทางกล้ามเนื้อใน CMT ชนิดที่ 1 [ 10 ]
การป้องกัน
การป้องกัน CMT สามารถทำได้โดยการปรึกษาทางพันธุกรรมกับพ่อแม่ในอนาคต โดยเฉพาะอย่างยิ่งหากคู่สามีภรรยามีประวัติครอบครัวเป็นโรคนี้ อย่างไรก็ตาม มีการระบุกรณีของการกลายพันธุ์ของยีนแบบ de novo point ซึ่งหมายความว่าในกรณีที่ไม่มีโรคนี้อยู่ในประวัติครอบครัว
ในระหว่างตั้งครรภ์ โอกาสเกิดโรค Charcot-Marie-Tooth ในลูกในอนาคตสามารถตรวจสอบได้ด้วยการตรวจชิ้นเนื้อรก (ตั้งแต่อายุครรภ์ 10 ถึง 13 สัปดาห์) รวมถึงการวิเคราะห์น้ำคร่ำ (เมื่ออายุครรภ์ 15 ถึง 18 สัปดาห์)
พยากรณ์
โดยทั่วไป การพยากรณ์โรค Charcot-Marie-Tooth แต่ละประเภทจะขึ้นอยู่กับความรุนแรงทางคลินิก แต่ในทุกกรณี โรคจะดำเนินไปอย่างช้าๆ ผู้ป่วยหลายรายมีอาการพิการ แม้ว่าจะไม่ทำให้มีอายุขัยสั้นลงก็ตาม